Los N-glicanos son complejos macromoleculares formados a partir de un precursor sintetizado en el retículo endoplásmico rugoso (RE). Consta de un oligosacárido ya preformado compuesto por N-acetil glucosamina, manosa y glucosa el cual se une a la asparagina de la proteína. El proceso de síntesis de los N-glicanos o N-glicosilación es, junto con la fosforilación, una de las modificaciones postrasduccionales más importantes que acontece en las proteínas (1-3). Este proceso, que conduce a la biosíntesis de los mencionados N-glicanos, implica varios pasos y se inicia, como hemos dicho, en el RE desde donde son transportados, mediante vesículas, al complejo de Golgi (CG). Ya en este orgánulo, los oligosacaridos, tanto de los lípidos como de las proteínas, son reordenados de forma muy precisa y específica. Posteriormente los glicolípidos y las glicoproteínas son empaquetados en vesículas y se transportan gracias al citoesqueleto y proteínas asociadas hasta su destino final, bien sea este su secreción, bien su incorporación a la membrana plasmática u otras endomembranas. Existen tres subtipos de N-glicanos: 1) los ricos en manosa, cuyas cadenas finalizan en manosa, 2) los híbridos que contienen cadenas con manosa terminal y otras que finalizan en ácido siálico y, 3) los complejos en los que todas las cadenas de los oligosacaridos presentan ácido siálico terminal. Por otra parte, la expresión y relativa abundancia de los N-glicanos está controlada por la acción secuencial y específica de varias glicosiltransferasas y glicosidadas (1-3). Los errores en los patrones de ramificación de los N-glicanos pueden causan múltiples anomalías denominadas “desórdenes congénitos en la glicosilación” (CDG), que incluyen importantes defectos neurológicos (4).
Los carbohidratos unidos a proteínas juegan una amplia variedad de papeles en la vida de la célula. Una de las funciones principales de estos glicanos es la de proveer epítopos de reconocimiento adicionales a los receptores de proteínas. Este sistema de reconocimiento esta implicado en multitud de procesos tales como el control de calidad y plegamiento de las proteínas, el tráfico o transporte de las mismas, la iniciación del proceso inflamatorio, la defensa del huésped, etc. Además, los carbohidratos complejos de la superfície celular juegan un papel relevante durante el desarrollo tal como se deduce por los profundos cambios que experimentan en su estructura de forma específica a lo largo del tiempo y del espacio durante la evolución de dicho proceso (5).
El desarrollo del sistema nervioso central (SNC) depende de que la migración neuronal y el crecimiento axonal se realicen según patrones muy estrictos estando, ambos procesos, regulados, entre otros factores, por interacciones intercelulares, en que intervienen no solo neuronas sino también células gliales. Estas interacciones están mediadas por las llamadas moléculas de adhesión celular, glicoproteínas que incluyen caderinas, selectinas, integrinas, mucinas y miembros de la superfamilia de las inmunoglobulinas (IgG). Entre estas últimas destacan la molécula de adhesión celular neural (NCAM) y la L1. Alteraciones en la expresión de estas moléculas conducen a graves defectos neurológicos (6).
El consumo de etanol durante la gestación puede inducir importantes efectos adversos sobre el desarrollo fetal, incluyendo obviamente el cerebro, hecho que puede llevar a la aparición de retraso mental en grados diversos (desórdenes en el desarrollo del SNC relacionados con el alcohol o ARND). En un extremo, se encuentra el síndrome alcohólico fetal (SAF) cuyo diagnóstico esta basado en tres criterios (a) deficiencias en el desarrollo, (b) desórdenes del SNC y, (c) un patrón distintivo de características faciales anormales (7). Los datos más fidedignos sobre su prevalencia señalan que la incidencia del SAF junto con ARND sería casi del 1 % de los nacimientos (8). Esta incidencia confirma la percepción de muchos especialistas de que la exposición prenatal al etanol es un problema grave y de que es la causa más importante de retraso mental en el mundo occidental. Además, el seguimiento de pacientes de distintas edades con estas alteraciones permite concluir que muchos de ellos tienen graves problemas de adaptación social y laboral.
A pesar de la simplicidad química del etanol, hoy en día no están claros los mecanismos por los que esta sustancia ejerce su acción teratogénica sobre el SNC y otros órganos habiéndose postulado diferentes hipótesis que confirman que el etanol tiene múltiples dianas en el SNC. Por ello, no sorprende que el alcohol dañe al feto a través de distintos mecanismos, entre los que se incluyen estrés oxidativo, interferencias con factores de diferenciación (retinol), inducción de apoptosis, supresión de la neurogénesis, disrupción de las interacciones intercelulares, alteraciones en la morfogénesis y en la liberación de distintos tipos de sustancias importantes para el correcto funcionamiento celular (6,9). Dentro de esta diversidad, una de los mecanismos propuestos por distintos laboratorios sugiere que el etanol altera la biosíntesis de glicanos y su posterior transporte intracelular. Este mecanismo se ha analizado principalmente en hepatocitos y en astrocitos aunque existe algún trabajo realizado también en neuronas. A continuación resumiremos los principales resultados de estos estudios llevados a cabo en astrocitos en fase de crecimiento o proliferación, ya que es durante esta etapa cuando los astrocitos producen una serie de factores como son, entre otros, la NCAM y el factor de crecimiento neural (NGF), capaces de interaccionar con las neuronas modulando su migración y posterior maduración durante el desarrollo del SNC (10, 11).
En distintos estudios llevados a cabo en hepatocitos de animales adultos expuestos de forma crónica o aguda al etanol se demostró que dicha exposición comporta la acumulación intracelular de proteínas, lípidos y consecuentemente agua, lo que resulta en el hinchamiento del hepatocito y finalmente a la aparición de hepatomegalia (12-15).
Al principio estos efectos se relacionaron con la posibilidad de que el acetaldehído, el primer metabolito del alcohol en hígado, formase aductos con distintas proteínas, como la tubulina no polimerizada e impidiese su correcta polimerización (16). La consecuencia es la formación de microtúbulos (MT) incompetentes para el transporte de las vesículas, lo que comportaría su retención o, al menos, un transporte mucho más lento. Un segundo mecanismo estaría relacionado con una menor eficiencia en la actividad enzimática de la glicosiltransferasas y glicosidadas. Ello resultaría, por una parte, en la eliminación/adición más lenta de los residuos azucarados y, por otra, en la eliminación/adición errónea de residuos lo que a su vez, produciría la aparición de “microheterogeneidad” en los glicanos de la superficie celular. Un ejemplo de este fenómeno lo tenemos en la “transferrina deficiente en carbohidratos (CDT)”, que es una variedad sérica de la transferrina, la cual se considera como uno de los marcadores más fiables en el consumo crónico de etanol (17).
En nuestro laboratorio hemos visto que en los hepatocitos expuestos prenatalmente al etanol se produce una retención significativa de glicoproteínas en el CG. Hay que destacar, no obstante, que dicha retención no afecta por igual a todas las proteínas susceptibles de ser transportadas (14,15).
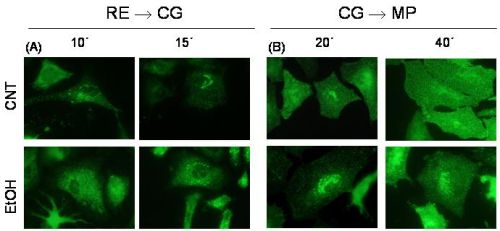 |
Figura 1. Cuando las células, una vez infectadas por un mutante termosensible del VSV, son expuestas a 40ºC (temperatura no permisiva) la glicoproteína VSV-G no puede plegarse correctamente y se retiene en el RE. A la temperatura permisiva de 32ºC, la proteína se pliega adecuadamente y se transporta hasta el CG lo que puede visualizarse mediante técnicas de inmunofluorescencia. De igual manera, para sincronizar la salida de la VSV-G desde este orgánulo, las células se someten a una temperatura de 20ºC, con lo que se produce la acumulación de la proteína en este punto. Al incubar nuevamente a 32ºC se produce la salida de la VSV-G desde el GC hasta la membrana plasmática. En esta figura se ilustra el efecto del etanol sobre el transporte del VSV-G. (A): En el transporte desde el RE al CG el etanol induce un retraso en la llegada de la VSV-G hasta el CG como se demuestra por el diferente patrón de fluorescencia, que en el caso de los controles (15 min) se corresponde al CG mientras que en el caso del etanol es todavía un patrón difuso característico del RE. (B): De igual manera el alcohol retrasa el transporte de VSV-G desde el CG hasta la membrana plasmática (MP). |
A nivel del SNC en los estudios sobre astrocitos en fase de proliferación hemos visto que el etanol induce la retención intracelular del NGF y de la forma inmadura polisializada de la NCAM (PSA-NCAM) (11, 18). Además, hemos podido demostrar que el alcohol induce la retención de proteínas víricas (p.e. glicoproteína del virus de la estomatitis vesicular o VSV-G) cuando las células son infectadas experimentalmente por estos virus (19). Por otra parte, el análisis del transporte intracelular de este virus demostró que la exposición al alcohol producía un enlentecimiento en todas las rutas de la vía secretora (RE-CG y CG-MP) (19) (Fig.1).
En base a estos resultados nos preguntamos si el retraso en el transporte de la VSV-G se debía a que el etanol alteraba la propia estructura del CG y/o los niveles de expresión de distintas proteínas implicadas en la funcionalidad de la vía secretora. Así, mediante técnicas de inmunofluorescencia utilizando marcadores del CG y mediante microscopia electrónica cualitativa y cuantitativa (estereología) vimos que efectivamente el etanol alteraba la morfología del CG traduciéndose en un hinchamiento de las cisternas y una reducción en el número de vesículas asociadas (19) (Fig.2). No obstante, estas alteraciones vistas en astrocitos son de menor envergadura que las observadas en hepatocitos (14, 20). El análisis del efecto del etanol sobre la maquinaria molecular implicada en la vía secretora mostró una respuesta heterogénea. Así, en relación con la superfamilia de proteínas denominadas SNARE e implicadas en el proceso de fusión de vesículas con la membrana adecuada (21), vimos que el etanol disminuía principalmente aquellas que participan en el transporte anterógrado que media entre el RE y el CG, concretamente las SNARE bet1 y sec22. El análisis de otro grupo de proteínas conocidas como Rab GTPasas, que también regulan la fidelidad del transporte y fusión/fisión de vesículas (22) dio como resultado que el etanol también disminuía algunas implicadas en el transporte anterógrado (Rab1, Rab2) y otras en el post-CG (Rab8). Por tanto, en conjunto, nuestros resultados concluyen que el etanol afecta tanto la integridad morfológica del CG (Fig.2) como a la maquinaria molecular implicada en el transporte secretor de proteínas, principalmente entre el RE y el CG.
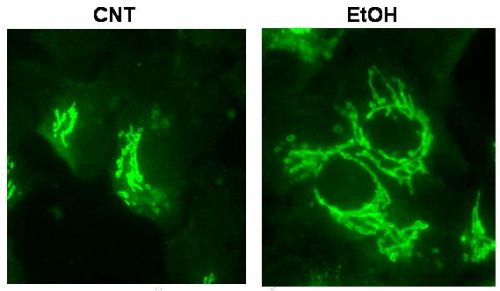 |
Figura 2. Imágenes obtenidas mediante fluorescencia utilizando un anticuerpo que marca la giantina, proteína marcadora del CG. Como se observa, la exposición al etanol (derecha) da como resultado que el CG ocupa una mayor área como consecuencia de un hinchamiento de las cisternas. |
Con el fin de examinar si el etanol alteraba la biosíntesis de los N-glicanos, los astrocitos se sometieron a pulsos de distinta duración (5 o 90 min) con diferentes precursores carbohidratos de la biosíntesis de N-glicanos marcados con tritio. Observamos que el etanol incrementaba la incorporación de la mayoría de los precursores estudiados y en particular la manosa (Man), la N-acetilmanosamina (ManNac), que es el precursor del ácido siálico y la 2-deoxy-glucosa (2-DGlc), que a su vez es el precursor no metabólico de la glucosa. En relación a la 2-DGlc vimos también que se acompañaba por un aumento en los niveles de GLUT1, el cual es el transportador específico de glucosa en astrocitos (Fig.3) (23, 24). Hay que destacar que dada la importancia del metabolismo de la glucosa en el cerebro, las variaciones en la captura de este compuesto en células nerviosas podrían tener una importante significación fisiológica.
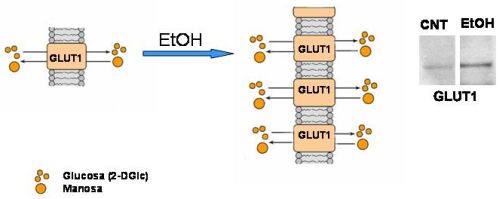 |
Figura 3. Esquema sobre la posible acción del etanol sobre el transporte de glucosa y de su transportador GLUT1. La exposición al etanol tiene como consecuencia un aumento en la captura de varios carbohidratos incluyendo glucosa y manosa que se acompaña con un incremento en la cantidad de GLUT1. |
Al mismo tiempo estudiamos la existencia de microheterogeneidad en las cadenas de N-glicanos ya que como hemos mencionado previamente, el etanol modifica tanto la cantidad como el tipo de glicanos en hepatocitos adultos y fetales. Para este estudio empleamos las lectinas, estas sustancias reconocen de forma más o menos específica monoglicanos o poliglicanos. Observamos que el etanol producía un aumento en los glicanos ricos en manosa. Otros grupos han demostrado también que el etanol, disminuye la actividad de diversas glicosiltransferasas y glicosidasas lo que contribuiría globalmente a la aparición de glicolípidos y glicoproteínas aberrantes en la superficie celular (23).
La endocitosis es un proceso mediante el cual las células ingieren compuestos del exterior que posteriormente envían a diversos compartimentos intracelulares. Con el fin de analizar si el etanol podía alterar este proceso analizamos su efecto sobre la internalización y posterior destino de dos proteínas, la transferrina y la albúmina, y de los glicoesfingolípidos esfingomielina y glucosilceramida (25, 26). La transferrina, una vez internalizada, es reciclada de nuevo al exterior mientras que la albúmina es transportada a los lisosomas para su posterior degradación. La exposición al etanol afectaba de distintas maneras la endocitosis de ambas proteínas.
El estudio de la internalización de los derivados lipídicos NBD-esfingomielina y NBD-glucosilceramida mostró que, al contrario de lo que ocurre en neuronas donde ambos compuestos se internalizan siguiendo una vía que finaliza en el CG, en la mayoría de los astrocitos la NBD-glucosilceramida seguía esta misma vía mientras que la NBD-esfingomielina se dirigía hasta los lisosomas. El etanol no variaba esta ruta pero producía un retraso considerable en la llegada de ambos compuestos lipídicos a sus destinos finales sin alterar la cantidad de esfingolípido endocitado.
Además de la maquinaria molecular de reconocimiento de membranas (SNAREs y Rabs), el transporte secretor y endocítico se sustenta en el citoesqueleto y proteínas asociadas tanto de MT como de filamentos de actina. En el caso del transporte vesicular los elementos que intervienen de forma más relevante son los MT que actúan como “autopistas” por donde circulan las vesículas en ambos sentidos con la ayuda de motores (dineína y kinesina) en función del sentido de la marcha (27,28). Los MT son polímeros polarizados compuestos por dímeros de isoformas de la tubulina. Son muy dinámicos ya que continuamente se ensamblan y se desensamblan. También los filamentos de actina son muy dinámicos en cuanto a su organización tridimensional y su papel en el transporte es menos conocido que el de los MT.
La pista de que los elementos del citoesqueleto se pudieran ver dañados por el etanol, dando como resultado alteraciones en el transporte vesicular, la dio el que los hepatocitos retenían en el CG proteínas tales como albúmina, así como que la tubulina no polimerizaba de forma eficaz, esto se atribuyó a la capacidad que tiene el acetaldehído, primer catabolito del etanol, para formar aductos con distintas proteínas, entre las que se encuentran la tubulina y la albúmina (12-15). Aunque en el SNC la formación de acetaldehído es menos relevante que en el hígado, podía ser suficiente para explicar el daño observado en la dinámica de los MT en astrocitos tratados con etanol (29). Esta dinámica la estudiamos analizando la cinética de despolimerización de los MT al tratarlos con nocodazol (NZ) así como la cinética de reensamblaje al retirar la droga. En estas condiciones observamos que tanto la repolimerización de los MT como la reestructuración del CG tras el tratamiento con NZ era más lenta en las células expuestas al etanol. Hay que señalar que a diferencia de lo que se ha descrito en hepatocitos la cantidad de tubulina no polimerizada no se veía alterada por el etanol. También vimos que el etanol producía un descenso en los niveles proteico de los motores de MT dineína y kinesina. Todos estos efectos eran dosis-dependientes (19, 24).
El segundo elemento del citoesqueleto que en astrocitos también se ve afectado por le etanol son los filamentos de actina. Mientras que en hepatocitos fetales no se observa ningún efecto morfológico del etanol sobre los mismos, en astrocitos se produce un cambio muy dramático en la típica organización en haces de las fibras estrés (24, 30). Los astrocitos tratados con etanol muestran una pérdida de las fibras de estrés y un reforzamiento periférico de la actina justo por debajo de la membrana plasmática (Fig.4). Este efecto es también dosis-dependiente. Hay que tener en cuenta la posibilidad de una conexión funcional entre los cambios en la reorganización de la actina cortical con un incremento de GLUT1 en la membrana plasmática y el consiguiente aumento de captura de glucosa por parte de los astrocitos tratados con etanol. Se sabe que el tratamiento con agentes que alteran la organización del citoesqueleto de actina comporta un incremento en la captura de glucosa del medio (31,32).
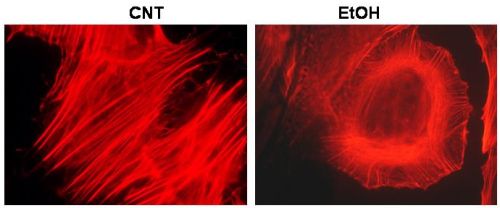 |
Figura 4. La exposición al etanol afecta de una forma dramática la organización de la actina polimerizada (fibras de estrés) tal como se revela mediante el marcado fluorescente con faloidina, un compuesto que se une de forma específica a la F-actina. Como se ve en la imagen las fibras pasan de disponerse paralelamente a reorganizarse en forma de anillo periférico. |
Viendo que el etanol produce una alteración en la reorganización de las fibras de estrés pensamos que tal vez la vía de señalización que regula su ensamblaje y que está gobernada por la proteína Rho estuviese dañada. De hecho, células que expresan un mutante constitutivamente activado de Rho A se vuelven resistentes a los daños producidos por el etanol sobre la actina y la captura de glucosa (24). Un elemento presente en el suero y que estimula la vía de la Rho GTPasa y el consiguiente ensamblaje de las fibras de estrés es el ácido lisofosfatídico (LPA). Observamos que este agente es capaz de revertir e incluso prevenir los daños del etanol sobre el citoesqueleto de actina y la captura exagerada de glucosa que se produce en los astrocitos. También normalizaba la cinética de reensamblaje de los MT. Estos resultados apuntan a las Rho GTPasas como dianas moleculares del etanol y definen al LPA como un posible agente citoprotector (24).
Los hallazgos encontrados en estos estudios apoyan la hipótesis de que las alteraciones inducidas por el etanol sobre la biosíntesis de los glicanos así como, sobre los sistemas de transporte intracelular, incluyendo el citoesqueleto, tienen una relevancia fundamental en el efecto teratogénico del etanol en los astrocitos y así, en el desarrollo del sistema nervioso.