Los protozoos parásitos son responsables de algunas de las enfermedades más comunes y devastadoras que afectan el hombre y a los animales domésticos, amenazando la vida de una tercera parte de la población mundial (34) . A pesar de su grave impacto, al afectar a los habitantes de las regiones más pobres del planeta, estas enfermedades quedan marginadas del interés de la industria farmacéutica. Todavía no se dispone de una vacuna efectiva para prevenirlas y, por lo tanto, la quimioterapia sigue siendo la principal forma de lucha contra los parásitos. Aunque estos organismos podrían ofrecer atractivas dianas terapéuticas gracias a sus rutas bioquímicas específicas, desafortunadamente la mayoría de los fármacos antiparasitarios llevan usándose desde hace más de cinco décadas. De hecho, según un análisis de Pecoul y col. (36) , de los 1233 fármacos nuevos comercializados entre 1975 y 1997, sólo 13 fueron aprobados para enfermedades tropicales. Además, muchos de estos fármacos antiparasitarios clásicos son altamente tóxicos y han dejado de ser efectivos debido a la aparición de resistencias en los parásitos, lo que se ha convertido en un importante problema de salud pública (58) . Entre estas enfermedades, la leishmaniasis es la que más muertes provoca tras la malaria (61) , por lo que es una de las enfermedades “olvidadas” que se han convertido en una prioridad para la Organización Mundial de la Salud (OMS) (61) .
La leishmaniasis es una enfermedad compleja (62) , causada por hasta 20 especies distintas del género Leishmania , un protozoo parásito perteneciente a la familia Trypanosomatidae . Se transmite por la picadura de un insecto vector, un mosquito de la subfamilia phlebotomina e. Leishmania presenta un ciclo de vida digenético (ver (63) ) que incluye una forma flagelada y móvil, llamada promastigote, que reside en el mosquito, y una forma inmóvil e intracelular, llamada amastigote, que se multiplica dentro de los macrófagos del individuo infectado, evadiendo de esta forma su sistema inmune. La enfermedad puede desarrollarse con un amplio abanico de manifestaciones clínicas de distinta gravedad, en función de la especie de Leishmania responsable de la infección y de las interacciones con el sistema inmune del individuo infectado. La forma mas grave de la enfermedad es la leishmaniasis visceral (LV) o Kala-azar (“fiebre negra” en hindú), mortal en ausencia de tratamiento. Se caracteriza por espasmos irregulares y fiebre, pérdida sustancial de peso, hinchazón del bazo y del hígado, y anemia. Los principales focos actuales se encuentran en Sudán y en la India. Menos grave es la leishmaniasis cutánea (LC), que se manifiesta en forma de numerosas úlceras en la piel, conocidas como “botón de oriente” o “úlcera de Bagdah”, en las partes expuestas del cuerpo como cara, brazos y piernas, causando una seria incapacidad y dejando al paciente con cicatrices permanentes. Estas lesiones suelen sanar por sí mismas. Actualmente, los principales focos de esta forma de leishmaniasis son Afganistán, Siria y Brasil. Por último, existen otras manifestaciones cutáneas de la enfermedad, destacando la leishmaniasis mucocutánea (LMC), que aparece en América latina y produce una destrucción extensiva y desfigurante de las membranas mucosas de la nariz, boca y garganta, la leishmaniasis difusa cutánea y la leishmaniasis dérmica post-kala-azar.
Desde 1993, las regiones endémicas para Leishmania se han expandido significativamente, acompañándose de un intenso incremento del número de casos de la enfermedad (21) . Se estima que existen 12 millones de enfermos, distribuidos en 88 países y aproximadamente 400 millones de personas expuestas, siendo la incidencia anual de nuevos casos de aproximadamente 2 millones (59) . Sin embargo, todos estos datos están probablemente subestimados, ya que la enfermedad es de comunicación obligada sólo en 32 de los 88 países que la padecen (59) . Además, no es un problema exclusivo de países tropicales, ya que el número de casos de LV en el litoral mediterráneo, y especialmente en España, también está aumentando hasta convertirse en un problema sanitario debido a la asociación a individuos con SIDA: se estima que en nuestro país la prevalencia de coinfección oscila entre 7-17% (1, 60) y se puede considerar como criterio diagnóstico de SIDA. Además, no existe un tratamiento eficaz frente a la LV en este tipo de inmunodeprimidos. Este hecho también se complica por la presencia en España de unos 560.000 perros, el reservorio natural de Leishmania spp, infectados por el parásito.
Ante la ausencia de vacunas efectivas y de un adecuado control del mosquito vector, la lucha contra la leishmaniasis recae en la quimioterapia. Sin embargo, el uso de los fármacos antimoniales de primera línea, que vienen usándose habitualmente desde hace más de 60 años y son muy tóxicos, está seriamente amenazado por el desarrollo de resistencia a los mismos (13) . De hecho, la leishmaniasis está incluida en la lista de las diez prioridades de lucha contra la resistencia a antibióticos por la OMS (58) . Además, parece cada vez más evidente que el éxito de los fármacos depende también de la variación en la sensibilidad de cada especie de Leishmania , de variaciones en su farmacocinética, y de variaciones en la interacción fármaco-respuesta inmune del paciente (13) . Afortunadamente, la situación actual de la quimioterapia de la leishmaniasis es más prometedora de lo que lo ha sido desde hace décadas, gracias a la reciente aparición de nuevos fármacos y de nuevas formulaciones de fármacos antiguos, ya aprobados o en fase clínica de estudio (ver las estructuras químicas en la Fig. 1).
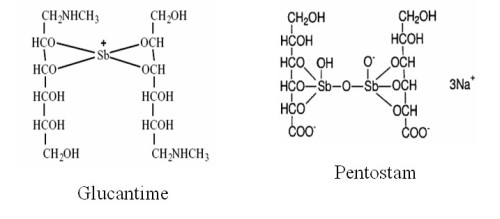 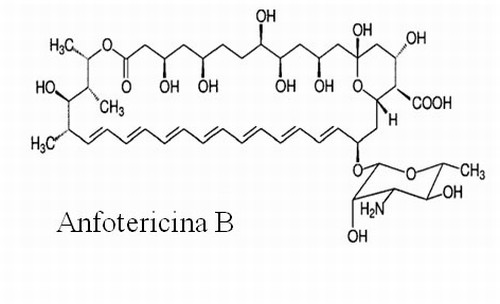 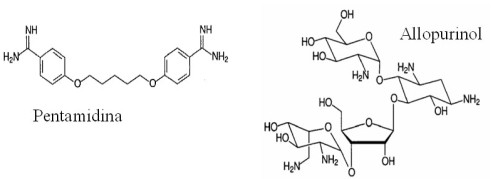 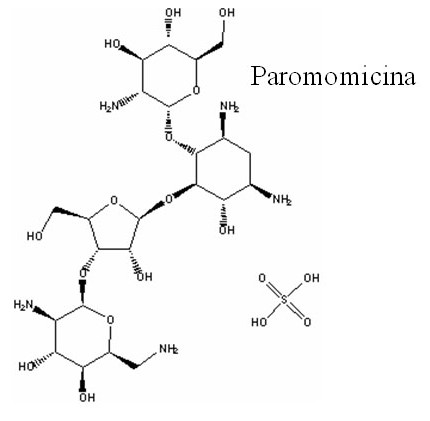   |
Figura 1. Fármacos usados actualmente para tratar la leishmaniasis |
El antimonio, un fármaco considerado como una de las siete maravillas del mundo en el siglo XVI, comenzó a utilizarse en su forma trivalente para tratar la leishmaniasis en 1913. Desde los años 40, los antimoniales pentavalentes en forma de Pentostam â (estibogluconato sódico) y Glucantime â (antimoniato de meglumina) constituyen la primera línea de fármacos recomendada por la OMS para todas las formas de leishmaniasis. Se administra intramuscular o intravenosamente durante 21-28 días, y producen efectos colaterales tóxicos como mialgias, pancreatitis, arritmias cardiacas y hepatitis, que en muchos casos llevan a la reducción o el cese del tratamiento (14) . Aunque su mecanismo de acción exacto sigue sin conocerse después de más de 60 años de uso, hoy día está aceptado que el antimonio pentavalente es un profármaco que tiene que reducirse a su forma trivalente para ejercer su acción. Sus dianas específicas no se han identificado, pero se sabe que inhiben la biosíntesis de macromoléculas en amastigotes, probablemente al perturbar el metabolismo energético debido a interferencias con la glucólisis y la b –oxidación de ácidos grasos (3) . También se conoce que su modo de acción involucra diversos efectos sobre el metabolismo del tripanotión (ver Fig. 2).
A pesar de que estos compuestos continúan siendo la primera línea de tratamiento en muchas partes del mundo, la aparición en los últimos 15 años de fenómenos de resistencia amenaza seriamente su uso. Por ejemplo, en ciertas zonas hiperendémicas de la India se produce hasta un 60% de fallo terapéutico (50) , probablemente debido al desarrollo de cepas de L. donovani resistentes a antimoniales (25) , por lo que ha dejado de ser el fármaco de elección. A su vez, en Irán recientemente se ha demostrado que el fallo terapéutico del glucantime para tratar la LC endémica se debe también a la aparición de líneas resistente a este fármaco (20) . La Fig. 2 representa los mecanismos moleculares propuestos para explicar la resistencia a antimoniales en Leishmania spp. (recientemente revisado en (13) ), entre los que destaca: i) una disminución de la reducción biológica del Sb V a Sb III (49) ; ii) una alteración en la expresión de la aquagliceroporina 1 (30) , un transportador responsable de la entrada de Sb III (19) ; iii) el incremento en los niveles de tripanotión (33) debido al aumento en la actividad de los enzimas limitantes en la síntesis de este tiol, la g -glutamilcisteinsintetasa y/o la ornitina decarboxilasa; iv) el secuestro intracelular del Sb III acomplejado con tioles, mediado por la sobreexpresión de PgpA (revisado en (23) ), un transportador perteneciente a la superfamilia ABC; y v) el eflujo del Sb III acomplejado con tioles mediado por un transportador aún desconocido. La mayoría de estos mecanismos han sido descritos experimentalmente in vitro , por lo que es muy importante estudiar si también son importantes en casos clínicos de resistencia.
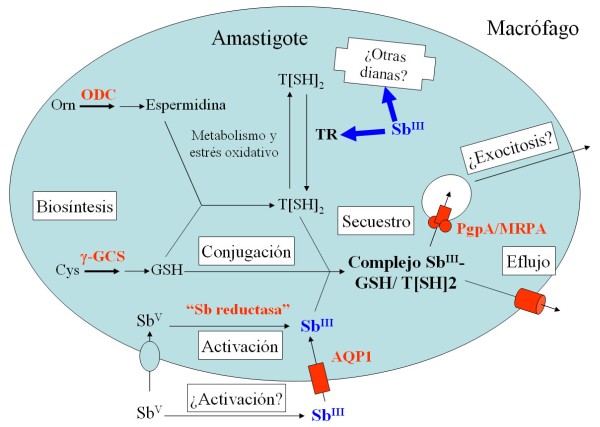 |
Figura 2: Mecanismo propuesto para explicar la acción del antimonio y la resistencia en Leishmania spp. Los niveles de ornitina descarboxilasa (ODC), -glutamilcisteinsintetasa ( -GCS) y glicoproteína-P-A (PgpA) están elevados en diversas líneas celulares resistentes, mientras que en otras líneas se observa una disminución de Sb reductasa. El SbIII inhibiría la tripanotión reductasa (TR) y otras dianas celulares. La internalización de SbIII está mediada por una aquaglicoporina (AQP1). Adaptado de (13) |
La anfotericina B es un antibiótico macrólido poliénico, predominantemente usado como antifúngico, que viene utilizándose como segunda línea de tratamiento de la leishmaniasis desde los años 60. Su mecanismo de acción se basa en su afinidad para unirse a esteroles y formar poros acuosos en la membrana de las células, a través de los cuales se pierden iones produciendo la muerte celular (5) . Su relativa especificidad sobre Leishmania spp. y hongos se debe a que se une con una ligera mayor afinidad por el ergosterol, el esterol predominante en estos microorganismos, que por el colesterol, presente en las células de mamíferos. Este bajo índice terapéutico explica los efectos tóxicos que produce en los pacientes, tales como cardiotoxicidad y nefrotoxicidad, que dificultan su uso (14) . Esta toxicidad se ha reducido considerablemente con la aparición de formulaciones lipídicas de la anfotericina B como el AmBisome â , Amphotec â y Abelcet â . La mayor limitación en este caso es su elevado coste, que lo hace prácticamente inaccesible en la mayoría de las zonas endémicas de la enfermedad.
La resistencia a anfotericina B no parece ser una amenaza para su uso clínico. In vitro , se ha conseguido seleccionar parásitos resistentes en los que se observó que el ergosterol de la membrana se sustituía por un precursor, probablemente por un defecto en su transmetilación (32) .
La paromomicina es un antibiótico aminoglicósido producido por Streptomices spp. Se usa para tratar la LV en una formulación parenteral, y la LC en una formulación tanto tópica como parenteral. Originalmente se desarrolló como agente antibacteriano, pero desde los años 60 se conoce su capacidad leishmanicida. Gracias al desarrollo de nuevas formulaciones del fármaco, se empezó a considerar como una alternativa eficaz para el tratamiento de la leishmaniasis a partir de los años 80. Su combinación con antimoniales se ha demostrado eficaz, describiéndose efectos sinérgicos in vitro , y aditivos en modelos animales (14) . Al contrario, su combinación con miltefosina produce efectos aditivos in vitro y sinérgicos in vivo (47) .
En cuanto a su mecanismo de acción frente a Leishmania , se sabe que la paromomicina altera la síntesis de proteínas. Además, interfiere con la actividad mitocondrial y altera la captación de precursores de macromoléculas por el parásito, lo que dificulta su crecimiento. Finalmente, incrementa la proporción de lípidos polares en la membrana, provocando una disminución de la fluidez que altera la permeabilidad de la membrana (28, 29) .
Aunque la resistencia clínica a aminoglicósidos en bacterias es bien conocida, el hecho de que no sea un fármaco de primera línea para tratar la leishmaniasis hace que no existan muchos casos clínicos de resistencia. En uno de ellos, se demostró que los parásitos procedentes de pacientes con recidivas fueron menos sensibles al fármaco que los aislados de los mismos pacientes antes de comenzar el tratamiento, sugiriendo que puede desarrollarse resistencia (53) . In vitro , es posible obtener poblaciones resistentes en las que se observa una reducción en la entrada del fármaco (27) . Dada la próxima introducción de la paromomicina para tratar la leishmaniasis, será importante definir mejor su mecanismo de acción y resistencia en el parásito (13) .
La pentamidina, una diamidina aromática, se ha utilizado como segunda línea de tratamiento para la leishmaniasis desde hace más de 40 años. Sin embargo, su uso no está muy extendido, probablemente debido a su toxicidad, ya que puede producir hipoglucemia, diabetes, nefrotoxicidad, taquicardia y dolor en el sitio de la inyección. Por el contrario, sí continúa administrándose para el tratamiento de la etapa temprana de la enfermedad del sueño, producida por Trypanosoma brucei , un parásito de la misma familia que Leishmania spp.
Su mecanismo de acción leishmanicida es debido a que se acumula con mayor selectividad en el parásito que en las células humanas (6) , y posiblemente incluye la inhibición del transporte de arginina, putrescina y espermidina, la unión al ADN del kinetoplasto provocando su desintegración, y al colapso del potencial de membrana mitocondrial (6, 13) .
Aunque el uso clínico limitado de la pentamidina hace poco probable el desarrollo de resistencias estables, in vitro si son muy fáciles de generar. Las líneas resistentes se caracterizan por mostrar una menor acumulación de fármaco (6) . La pentamidina entra al parásito, tanto sensible como resistente, por vía del mismo transportador específico localizado en la membrana plasmática, que no se encuentra alterado en las leishmanias resistentes. Sin embargo, en las líneas salvajes la pentamidina citoplasmática se acumula rápidamente en la mitocondria, gracias a su potencial de membrana, donde ejerce su efecto tóxico. Por el contrario, en las líneas resistentes, que presentan un potencial de membrana mitocondrial disminuido, el fármaco no se acumula tan rápidamente en la mitocondria, quedando libre en el citosol. Desde aquí, sería eliminado por medio de bombas activas, probablemente miembros de la familia ABC como PRP1 (9) , explicándose así la menor acumulación neta de fármaco observada en estas líneas. Por lo tanto, fármacos que inhiban este eflujo podrían revertir la resistencia al permitir la acumulación citosólica de pentamidina a niveles tales que conducirían a su acumulación mitocondrial (6) .
La capacidad leishmanicida de los azoles como el ketoconazol, el fluconazol y el itraconazol, originariamente desarrollados como antifúngicos, se conoce desde principios de los años 80. A pesar de que en la siguiente década se los estudió como una posible primera terapia oral y de que se conoce bien su mecanismo de acción, no ha habido demasiados avances en su uso terapéutico.
La diana de estos compuestos es la ruta biosintética del ergosterol, el esterol mayoritario en hongos y Leishmania spp. frente al colesterol mayoritario en las células de mamífero. Probablemente, la diana específica sea la C14 a -desmetilasa, como ocurre en hongos. La reducción en el nivel de esteroles normales y la acumulación de intermediarios anormales trastorna la membrana y diversas funciones metabólicas que conllevan a la muerte del parásito. Sin embargo, ante la presión de estos fármacos, Leishmania puede llegar a utilizar el colesterol del medio o de las células del hospedador para satisfacer muchos de los requerimientos celulares, lo que junto a la variabilidad en la sensibilidad según la especie de Leishmania , puede explicar los problemas encontrados en los estudios clínicos (14) .
No existen estudios de resistencia in vitro en Leishmania , aunque en el parásito Trypanosoma cruzi es muy fácil de inducir. En hongos, se ha visto que casos clínicos de resistencia se correlacionan con un incremento en el eflujo de fármaco mediado por transportadores ABC (13) .
El allopurinol, un análogo de la purina, entró en ensayos clínicos para tratar la LV y la LC en 1981. Aunque recientemente se ha demostrado que no es muy efectivo en humanos, continúa utilizándose para tratar la leishmaniasis canina. Esto es debido a que en humanos se produce un rápido metabolismo y excreción del fármaco que evita que se consigan niveles plasmáticos suficientes. Por el contrario, en el perro la farmacocinética del fármaco es diferente y si se obtienen fácilmente niveles plasmáticos leishmanicidas (13, 14) . Además, en humanos si podría tener una aplicación en combinación con otros fármacos, especialmente los antimoniales.
El modo de acción del allopurinol se basa en la incapacidad de Leishmania spp. para sintetizar purinas de novo , por lo que depende de rutas de salvamento para obtenerlas del hospedador. Los análogos de nucleósidos como el allopurinol se convierten probablemente en análogos de ribonucleósidos trifosfato que se incorporarían al ARN, alterando la síntesis de proteínas (31) . No hay descrito ningún caso de resistencia, ni experimental ni clínica.
La sitamaquina, una 8-aminoquinoleína sintetizada en 1944 y anteriormente conocida como WR6026, está actualmente en fase clínica II de estudio para el tratamiento de la LV en la India y Kenia (22, 57) . Este fármaco es un análogo de la primaquina, usada desde hace 50 años para tratar la malaria (2) , y presenta un amplio espectro de actividad antiprotozoaria (64) . Su mayor atractivo radica en su eficacia por vía oral, por lo que está en desarrollo por GlaxoSmithKline. Aunque en general es bien tolerada, la sitamaquina puede producir vómitos, dolor abdominal, dolor de cabeza, y en algunos casos, efectos renales severos que obligan a continuar con la investigación (22, 57) . Estos efectos renales no se habían visto en modelos animales. Esta diferencia podría ser debida a que la sitamaquina es biotransformada de distinta forma en los microsomas hepáticos humanos frente a los de otros mamíferos, y son estos metabolitos los que podrían jugar un papel importante tanto en la actividad leishmanicida como en los daños colaterales.
Su mecanismo de acción no es bien conocido. Se ha descrito que la sitamaquina provoca un rápido colapso del potencial de membrana interno de la mitocondria (54) , y la alcalinización de los acidocalcisomas (55) , unas vacuolas acídicas presentes en todos los tripanosomátidos que contienen la mayoría del calcio intracelular. Por el momento, no se ha descrito ningún caso de resistencia in vivo o in vitro , y resulta difícil de generarla experimentalmente (L Carvalho y col. comunicación personal).
La miltefosina (hexadecilfosfocolina) es el último fármaco leishmanicida en ser aprobado (2002), y el primero efectivo por vía oral. Originariamente se desarrolló como agente antitumoral, pero desde finales de los 80 se conoce su actividad leishmanicida, tanto in vitro como in vivo (12) . Su rápido desarrollo desde el laboratorio hasta su registro, en sólo 6 años, ha sido posible gracias a la colaboración entre el gobierno de la India, la compañía farmaceútica Zentaris y el consorcio internacional TDR (Tropical Diseases Research). En la India se ha registrado con el nombre de Impavido â , y se confía en que contribuya a erradicar la LV, endémica en la zona y con un alto nivel de resistencias a antimoniales. Además se ha registrado en Alemania y Colombia, en este último caso para tratar la LC. La miltefosina induce una rápida cura clínica y parasitológica (95% y 91% para LV y LC respectivamente), con dosis de 100-150 mg/día (ó 2,5 mg/kg de peso) durante 28 días. Tiene una larga vida media en el cuerpo, que varía entre 150 y 200 h, requiriéndose unas 4 vidas medias (25-33 días) para llegar a eliminar el 90% de los niveles plasmáticos. Por lo tanto, durante el curso de un tratamiento convencional, durante varias semanas quedan niveles subterapéuticos del fármaco, lo que puede provocar la aparición de resistencias en el caso de reinfección (7) . Aunque el índice terapéutico de la miltefosina es bajo, a las concentraciones efectivas es bien tolerado en general, incluyendo niños, por lo que se puede recomendar su elección como primera línea de tratamiento de la VL infantil en la India, que constituye la mitad de los casos (4, 51) . Las reacciones adversas más frecuentes son: molestias gastrointestinales transitorias, vómitos, diarrea, y aumento de las enzimas hepáticas y de la creatinina sérica. Normalmente estos problemas son suaves o moderados y no requieren la parada del tratamiento o la disminución de la dosis. Sin embargo, la miltefosina es teratogénica en animales, por lo que no puede administrarse en mujeres embarazadas y requiere el uso de anticonceptivos durante un periodo de 8 vidas medias de la miltefosina (2-3 meses) en mujeres con posibilidad de embarazo (52) .
Se ha demostrado que la eficacia de la miltefosina depende de la especie de Leishmania , siendo L. donovani la más sensible y L. major (17) o L. brazilensis (FJ Pérez-Victoria y col. resultados sin publicar) las menos. Esta variabilidad no se debe a resistencias adquiridas, sino que refleja diferencias en la susceptibilidad intrínseca al fármaco, lo que puede tener un importante impacto en el resultado clínico. Como la acumulación intracelular del fármaco es un prerrequisito para su acción leishmanicida, las diferencias de sensibilidad probablemente se deben a distinto niveles de expresión de la maquinaria necesaria para su internalización (ver más adelante).
El mecanismo de acción de la miltefosina no se conoce exactamente. Se ha descrito una perturbación del metabolismo de alquil-lípidos (26) , pero sólo a concentraciones mucho mayores de las necesarias para matar al parásito. También se ha descrito daño en la membrana flagelar y defectos en la síntesis de fosfolípidos (24, 46) . En cualquier caso, independientemente de cual sea su diana primaria, la miltefosina induce una muerte celular tipo apoptosis, con condensación y fraccionamiento del ADN, exposición de fosfatidil-serina y compactación celular (35, 56) . Probablemente la miltefosina produce diferentes efectos debido a su actividad detergente, que conduce a esta muerte tipo apoptosis. Por lo tanto, el requisito básico para su actividad leishmanicida y su especificidad para el parásito radica en su internalización. Además, como veremos en el siguiente apartado, el parásito es capaz de adquirir fácilmente resistencia a la miltefosina impidiendo su acumulación intracelular, por lo que este proceso ha sido muy estudiado en los últimos años. Se pueden distinguir tres etapas en la acumulación intracelular de miltefosina (39) i) la unión a la cara externa de la membrana plasmática, a partir de la miltefosina unida a la albúmina, que actúa como reservorio del fármaco; ii) la internalización, que no se produce por endocitosis ni difusión, sino por una actividad flipasa que transloca el fármaco a la cara interna de la membrana plasmática. Recientemente se ha descrito la maquinaria responsable de esta actividad (ver más adelante); y iii) la redistribución de la miltefosina en membranas intracelulares. En oposición, la miltefosina puede salir del parásito por exocitosis o por una actividad flopasa que la transloque desde la cara interna a la externa de la membrana plasmática, probablemente mediada por transportadores ABC.
Dada la reciente aprobación de la miltefosina para tratar la LV en la India y la LC en Colombia, todavía no se han encontrado aislados clínicos resistentes. Sin embargo, experimentalmente es muy fácil obtener parásitos resistentes a miltefosina, tanto por presión creciente con miltefosina (48) u otros fármacos (43) como por mutagénesis (38, 48) . En todos los casos descritos hasta el momento, la resistencia se debe a una menor acumulación intracelular de miltefosina, que puede deberse a un mayor eflujo del fármaco o a un impedimento de su captación (Fig. 3).
El primer caso de resistencia a miltefosina fue descrito en el año 2001 en una línea multirresistente (MDR) de L. tropica (43) . Esta línea resistente, que había sido seleccionada mediante adaptación a concentraciones crecientes de daunomicina, contiene una amplificación y sobreexpresión de un gen con alta similitud al de la glicoproteína-P (Pgp) humana (mdr1) (8) . Los parásitos presentan una resistencia cruzada a diversos fármacos de estructura química y mecanismo de acción muy diferente, de ahí el nombre de multirresistentes. La LPgp(LMDR1) de Leishmania spp. es una proteína de membrana de unos 150 kDa. Pertenece a la superfamilia ABC (por A TP B inding C assette), al igual que otros transportadores implicados en resistencia a agentes leishmanicidas como antimoniales, pentamidina y azoles (ver anteriormente) y a fármacos antimaláricos como mefloquina, cloroquina y artemisinina (16) . La Pgp utiliza la energía proveniente de la hidrólisis del ATP para expulsar todos estos fármacos fuera de la célula. Está compuesta por dos mitades homólogas, cada una de las cuales contiene un dominio transmembrana (TMD), involucrado en la unión y transporte del fármaco, y un dominio citosólico (NBD), que une e hidroliza el ATP, proporcionando la energía necesaria para el proceso de transporte.
Los parásitos MDR son altamente resistentes a miltefosina (entre 9 y 18 veces) ( (43) y JM Pérez-Victoria y col., resultados sin publicar), y también lo son frente al fármaco relacionado edelfosina. Esta resistencia es debida a un mayor eflujo de miltefosina en la línea MDR, que se traduce en una menor acumulación intracelular (Fig 3) (41) . La LPgp(LMDR1) es la responsable de esta resistencia actuando como flopasa de miltefosina (43) , ya que: i) el fenotipo de resistencia depende del nivel de sobreexpresión de la proteína; ii) la inhibición de la LPgp(LMDR1), como veremos más adelante, restaura la acumulación de miltefosina y la sensibilidad al fármaco; y iii) la miltefosina es capaz de modular la acumulación de daunomicina en la línea MDR, también dependiente de la sobreexpresión de este transportador. La localización de la LPgp(LMDR1) no está definitivamente establecida; aunque podría estar en la membrana plasmática, como el homólogo humano, y expulsar directamente el fármaco al exterior, recientemente se ha descrito una localización en vesículas intracelulares (15) . En este caso, la expulsión del fármaco intracelular se produciría por la exocitosis de estas vesículas que secuestrarían la miltefosina (Fig 4).
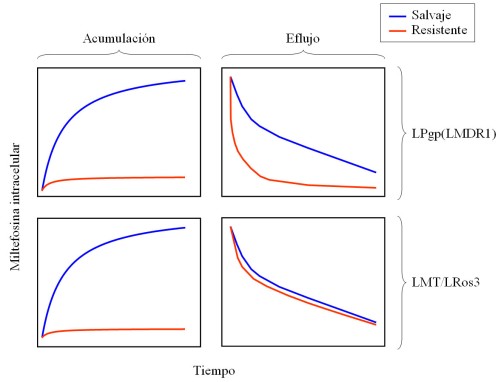 |
Figura 3. Acumulación y eflujo de miltefosina en líneas resistentes de Leishmania. Utilizando fármaco radioactivo se ha podido comprobar que la resistencia experimental a miltefosina en Leishmania spp. se explica, en todos los casos descritos, por una menor acumulación intracelular del fármaco. Esto puede deberse a un eflujo de la miltefosina mediado por la glicoproteína-P (LPgp(LMDR1)) o a un defecto en la maquinaria que permite su internalización (LMT/LRos3). |
La caracterización de todas las líneas de L. donovani seleccionadas hasta la fecha en presencia de altas concentraciones de miltefosina han mostrado siempre un defecto en la acumulación del fármaco, que no llega al 3% de los niveles observados en los parásitos controles (Fig. 3) (37) . Ni la actividad endocítica ni el eflujo y el metabolismo del fármaco están alterados en estas líneas, por lo que el mecanismo de resistencia, de hasta 15 veces (38, 48) , se basa en la internalización de la miltefosina (37) , es decir, en la actividad flipasa comentada anteriormente, capaz de translocar los monómeros de miltefosina desde la cara externa de la membrana plasmática a la interna. Estudios de complementación funcional usando estas líneas resistentes han permitido elucidar la maquinaria molecular responsable de esta actividad (revisado recientemente en (39) ): el transportador de miltefosina LdMT (38) y su unidad beta específica LdRos3 (Fig. 4).
El transportador de miltefosina LdMT es una proteína de membrana de 1097 aminoácidos perteneciente a la familia de ATPasas de tipo-P de aminofosfolípidos translocasas. Su eliminación por generación de mutantes nulos o la adquisición de mutaciones puntuales que inactiven su función impide la acumulación de miltefosina dentro de los parásitos, haciéndolos altamente resistentes (38) . Para poder realizar su función, LdMT debe de localizarse en la membrana plasmática. Es de destacar que la localización de LdMT, y por tanto su actividad, depende de la presencia de una subunidad beta específica, llamada LdRos3 (FJ Pérez-Victoria y col., manuscrito en preparación). Esta proteína de 366 aminoácidos pertenece a la familia CDC50/Lem, y contiene dos segmentos transmembrana separados por un largo bucle extracelular. Su correcta localización en la membrana plasmática depende a su vez de la presencia de una forma activa de LdMT, por lo que ambas proteínas son mutuamente dependientes para su localización y función. Cuando se elimina o inactiva cualquiera de las dos, el resultado es el mismo: la adquisición de altos niveles de resistencia a miltefosina. Todo esto sugiere que ambas proteínas forman parte de la misma maquinaria de translocación de miltefosina presente en la membrana plasmática del parásito. ¿Podría el mal funcionamiento de esta maquinaria originar casos clínicos de resistencia? Todavía no tenemos una respuesta a esta pregunta, que probablemente dependa del papel fisiológico que desempeñen estas proteínas in vivo , relacionado quizás con el mantenimiento de la asimetría de la membrana merced a su actividad translocadora de fosfolípidos (37) .
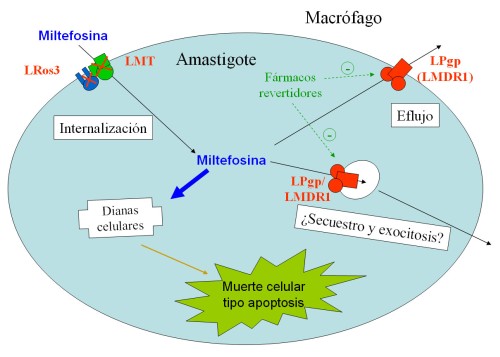 |
Figura 4. Mecanismo propuesto para explicar la acción de la miltefosina y la resistencia en Leishmania spp. La acción leishmanicida de la miltefosina, que resulta en una muerte de tipo apoptosis, es consecuencia de su acumulación específica en el interior del parásito. El fármaco entra a través de la maquinaria formada por el transportador de miltefosina (LMT) y su unidad beta específica (LRos3). El malfuncionamiento de esta maquinaria de internalización y/o la sobrexpresión de la glicoproteína-P (LPgp/LMDR1), que expulsa la miltefosina fuera del parásito, confiere altos niveles de resistencia al fármaco. La inhibición de la LPgp(LMDR1) (verde) impide este eflujo de miltefosina y sensibiliza a los parásitos resistentes. |
Con el registro de la miltefosina en la India, su Gobierno confía en eliminar la enfermedad en los próximos 10 años (18) . Sin embargo, varios factores sugieren desgraciadamente que estas previsiones pueden ser demasiado optimistas. Como hemos visto anteriormente, la larga vida media del fármaco y la necesidad de un tratamiento largo provoca que se mantengan niveles de miltefosina subterapéuticos durante varias semanas. Además, los parásitos pueden hacerse fácilmente resistentes ante la presión de fármaco disminuyendo su acumulación intracelular. A estos factores hay que sumarle el alto precio de la miltefosina, que llega a los 150 US$, claramente inasequible para la mayoría de la población de estas zonas endémicas en las que los ingresos diarios medios de una familia son de aproximadamente 1 US$. Estos precios, junto a la posibilidad de comprar y vender libremente pequeñas cantidades de fármaco, provocan que la duración del tratamiento no sea el adecuado. Por lo tanto, el uso futuro de la miltefosina como único fármaco en la India podría conducir a una rápida aparición de resistencia generalizada, lo que sería una tragedia que hay que evitar mediante: i) una política adecuada que asegure el buen uso del fármaco; ii) la combinación de la miltefosina con otros fármacos leishmanicidas; y iii) la reversión de la resistencia en el parásito.
El acceso gratuito al fármaco a través del sistema publico de salud en áreas endémicas, junto con una prescripción y seguimiento por médicos cualificados evitarían los factores extrínsecos al parásito que posibilitan la aparición de resistencias (39) . Además, es aconsejable que no se le dé una aplicación veterinaria a este fármaco.
La combinación de fármacos leishmanicidas para controlar el problema de la resistencia a fármacos es una de las estrategias a priori más importantes, y ya está adoptada para el tratamiento de otras enfermedades infecciosas. Los fármacos actuales contra Leishmania spp., como hemos visto, tienen estructuras químicas y mecanismos de acción muy diferentes, por lo que es menos probable que se generen resistencias (salvo en el caso de que los fármacos usados en la terapia combinatorial fueran sustratos de bombas de eflujo multiespecíficas como la LPgp(LMDR1)). Además, en algunos casos se ha descrito un sinergismo en la acción conjunta de distintos leishmanicidas, por lo que se podría disminuir la duración o la dosis del tratamiento, lo que también ayudaría a evitar la aparición de resistencias. Obviamente, mientras mayor sea el repertorio de fármacos leishmanicidas con diferente mecanismo de acción, mayor será la posibilidad de sortear la resistencia a fármacos (13) .
Recientemente, Seifert y Croft han estudiado las interacciones de la miltefosina con otros fármacos leishmanicidas, tanto in vitro como in vivo (47) . En un modelo de amastigotes intracelulares, demostraron sinergismo entre la miltefosina y los antimoniales, mientras que la interacción se describió como indiferente al combinar la miltefosina con anfotericina B, sitamaquina o paromomicina. Sin embargo, en modelos murinos el resultado fue diferente: no se encontró efecto sinérgico con los antimoniales pero sí con la anfotericina B y la paromomicina (la sitamaquina no fue incluida en los ensayos in vivo ). En función de la toxicidad y el coste del fármaco “pareja”, los autores aconsejan seguir con el estudio de la combinación de miltefosina y paromomicina. En otro parásito de la familia ( T. cruzi ), la miltefosina ha demostrado in vitro un efecto sinérgico con el ketoconazol, tanto en formas epimastigotes como en formas amastigotes (45) .
Como hemos visto anteriormente, Leishmania puede volverse resistente a la miltefosina mediante la sobreexpresión de la bomba de eflujo LPgp(LMDR1), que evita su acumulación intracelular. Por lo tanto, una forma eficaz de revertir este tipo de resistencia (aunque no la mediada por el malfuncionamiento de la maquinaria de internalización) es la inhibición específica de este transportador. Esta estrategia viene estudiándose desde hace más de 30 años para revertir la multirresistencia a fármacos mediada por la Pgp humana durante el tratamiento del cáncer, o más recientemente, para alterar la farmacocinética de fármacos sustratos de la bomba. Sin embargo, los inhibidores clásicos de la Pgp humana como el verapamilo o la ciclosporina A, que se unen a los dominios transmembrana del transportador, no son eficaces con el transportador de Leishmania (8) .
Durante los últimos años, se han desarrollado diferentes estrategias para inhibir la Pgp de Leishmania (42) . En primer lugar, se eligieron los dominios citosólicos de unión a nucleótidos (NBD) como nuevas dianas farmacológicas. Su purificación posibilitó la selección de inhibidores específicos, entre los que destacan los flavonoides, y el establecimiento de relaciones estructura-actividad que han permitido el diseño racional de inhibidores cada vez más potentes. Hay que destacar que existe una correlación total entre la afinidad de estos compuestos por los dominios citosólicos y su capacidad para modular la acumulación intracelular de fármaco y sensibilizar a los parásitos resistentes (10, 40, 43) . También se han seleccionado compuestos dirigidos frente a los dominios transmembrana (TMD) del transp ortador, destacando los sesquiterpenos con un esqueleto dihidro- b -agarofurano (11, 44) . Para estos compuesto s se han obtenido modelos cuantitativos de estructura-actividad (3D-QSAR) que han permitido conocer los requisitos químicos necesarios para su capacidad inhibidora (11) . Finalmente, se ha demostrado que la administración de subdosis de inhibidores dirigidos frente a las dos dianas dentro del transportador, NBD y TMD, revierte eficazmente la resistencia a miltefosina en los parásitos MDR al modular la acumulación de fármaco (Fig. 5) (41) .
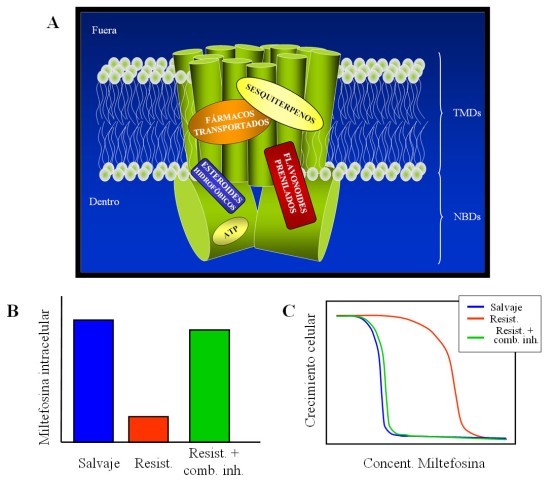 |
Figura 5. Reversión de la resistencia a miltefosina mediada por la sobreexpresión de la Pgp. A) Modelo propuesto para explicar la estrategia combinatorial seguida para inhibir la LPgp(LMDR1) de Leishmania. Los dominios citosólicos de la Pgp contienen, además del sitio de unión del ATP, un sitio de unión a compuestos hidrofóbicos. Los flavonoides prenilados se fijarían sobre los NBDs, habiéndose descrito su competición con nucleótidos y esteroides hidrofóbicos. A su vez, los sesquiterpenos con esqueleto dihidro--agarofurano se fijarían sobre los dominios transmembrana, aunque probablemente no directamente sobre los sitios de unión a fármacos. B) Modulación de la acumulación intracelular de miltefosina por la combinación de subdosis de inhibidores. C) Reversión de la resistencia a miltefosina por la combinación de subdosis de inhibidores |
Durante más de 60 años, los antimoniales pentavalentes han sido, a pesar de todos sus defectos, los fármacos elegidos para tratar la LV y la LC. Nuevas alternativas como la anfotericina B liposomal, la paromomicina y posiblemente la sitamaquina han despertado gran interés, aunque el precio, la producción de formulaciones comerciales o los efectos secundarios siguen siendo un problema. Sin embargo, ha sido el reciente registro de la miltefosina como el primer fármaco oral para el tratamiento de la LV y LC lo que ha abierto las mayores expectativas de eliminar la enfermedad en zonas hiperendémicas. No obstante, la farmacocinética del fármaco, su precio y la facilidad que tiene el parásito para volverse insensible aconsejan el establecimiento de políticas que prevengan la aparición de estas resistencias. El descubrimiento de los mecanismos responsables de la resistencia experimental a miltefosina, como son su eflujo activo a través de LPgp/LMDR1 y la alteración de su internalización a través de la maquinaria LdMT/LdRos3, puede ayudar a desarrollar marcadores que permitan supervisar la aparición clínica de resistencias, así como sensibilizar a los parásitos resistentes debido a la sobreexpresión de la LPgp mediante el empleo de agentes revertidores. En todo caso, la experiencia nos obliga a no bajar la guardia y a continuar el esfuerzo en la búsqueda de nuevas dianas que permitan el desarrollo de nuevos fármacos leishmanicidas más efectivos, seguros, baratos, y a ser posible, de administración oral.
Los autores agradecen a la Red del FIS RICET C03-04, al Plan Andaluz de Investigación de la Junta de Andalucia (Cod. Grupo CI130), al Proyecto de la UE Referencia QLRT-2000-01404 y a la Compañia Farmacéutica Zentaris (Frankfurt, Alemania) por la financiación e interés durante el desarrollo de los experimentos dirigidos a conocer los mecanismos de resistencia a miltefosina en Leishmania .